Affiliated scientific publications
2023
- Katsiari, M., Mavroidi, A., Kesesidis, N., Palla, E., Zourla, K., Ntorlis, K., Konstantinidis, K., et al. (2023). ‘Emergence of Clonally-Related South Asian Clade I Clinical Isolates of Candida auris in a Greek COVID-19 Intensive Care Unit’, Journal of Fungi, 9/2. MDPI. DOI: 10.3390/jof9020243
- Mpekoulis, G., Kalliampakou, K. I., Milona, R. S., Lagou, D., Ioannidis, A., Jahaj, E., Chasapis, C. T., et al. (2023). ‘Significance of Catecholamine Biosynthetic/Metabolic Pathway in SARS-CoV-2 Infection and COVID-19 Severity’, Cells, 12/1. MDPI. DOI: 10.3390/cells12010012
- Pliakou, E., Lampropoulou, D. I., Dovrolis, N., Chrysikos, D., Filippou, D., Papadimitriou, C., Vezakis, A., et al. (2023). ‘Circulating miRNA Expression Profiles and Machine Learning Models in Association with Response to Irinotecan-Based Treatment in Metastatic Colorectal Cancer’, International Journal of Molecular Sciences, 24/1. MDPI. DOI: 10.3390/ijms24010046
- Rezasoltani, S., Aghdaei, H. A., Jasemi, S., Gazouli, M., Dovrolis, N., Sadeghi, A., Schlüter, H., et al. (2023). ‘Oral Microbiota as Novel Biomarkers for Colorectal Cancer Screening’, Cancers, 15/1. MDPI. DOI: 10.3390/cancers15010192
2022
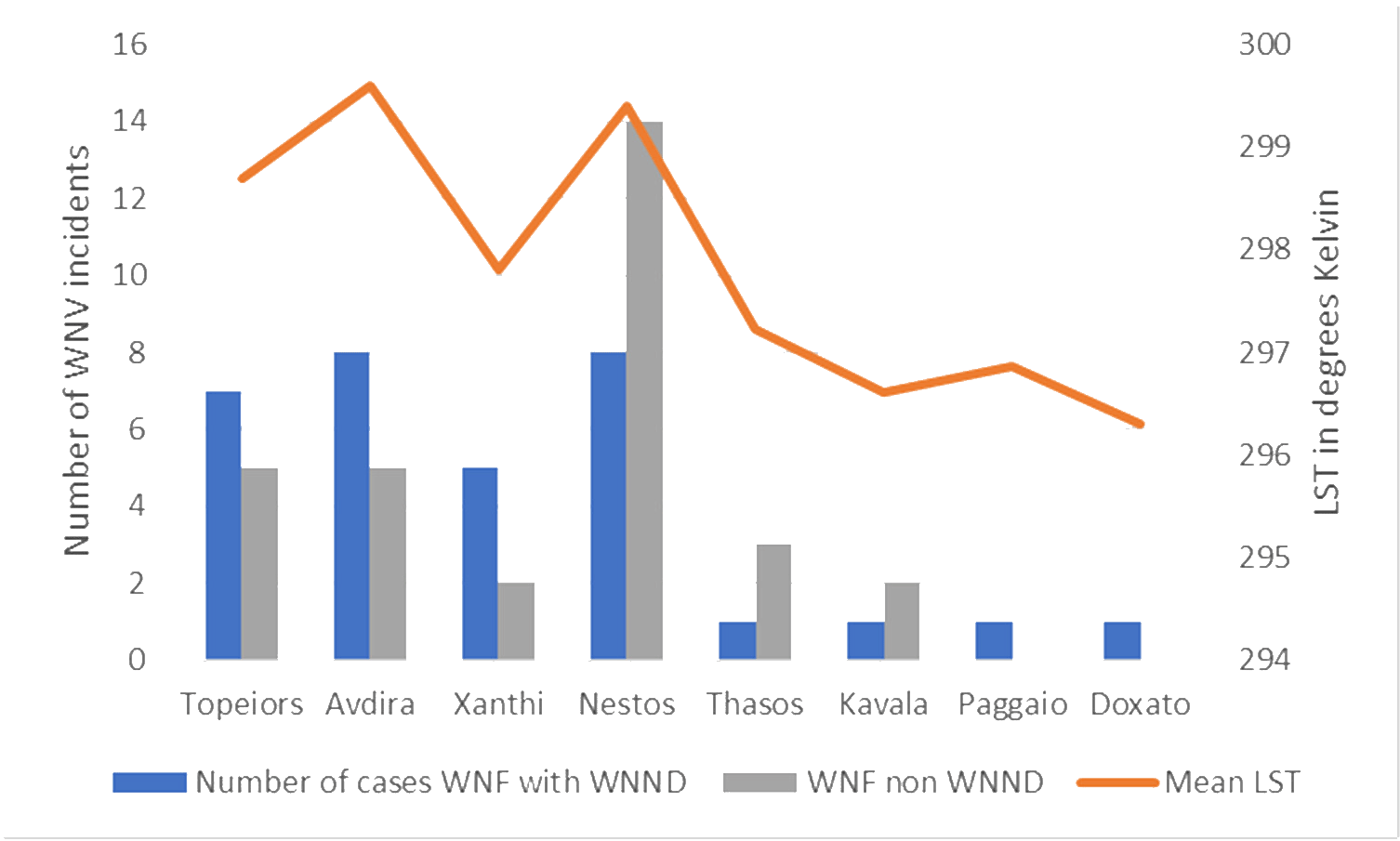
- Bampali, M., Konstantinidis, K., Kellis, E. E., Pouni, T., Mitroulis, I., Kottaridi, C., Mathioudakis, A. G., et al. (2022). ‘West Nile Disease Symptoms and Comorbidities: A Systematic Review and Analysis of Cases’, Tropical Medicine and Infectious Disease, 7/9. MDPI. DOI: 10.3390/tropicalmed7090236
- Beka, A. A., Papadopoulos, V., Mylopoulou, T., Panopoulou, M., Karakasiliotis, I., Mavromara, P., Mimidis, K., et al. (2022). ‘Association between vitamin D receptor gene polymorphisms and fibrosis susceptibility in Greek patients with HCV infection’, GERMS, 12/3: 384–93. European Academy of HIV/AIDS and Infectious Diseases. DOI: 10.18683/germs.2022.1342
- Dovrolis, N., Filidou, E., Tarapatzi, G., Kokkotis, G., Spathakis, M., Kandilogiannakis, L., Drygiannakis, I., et al. (2022). ‘Co-expression of fibrotic genes in inflammatory bowel disease; A localized event?’, Frontiers in Immunology, 13. Frontiers Media S.A. DOI: 10.3389/fimmu.2022.1058237
- Evangelou, K., Veroutis, D., Paschalaki, K., Foukas, P. G., Lagopati, N., Dimitriou, M., Papaspyropoulos, A., et al. (2022). ‘Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis’, European Respiratory Journal, 60/2. European Respiratory Society. DOI: 10.1183/13993003.02951-2021
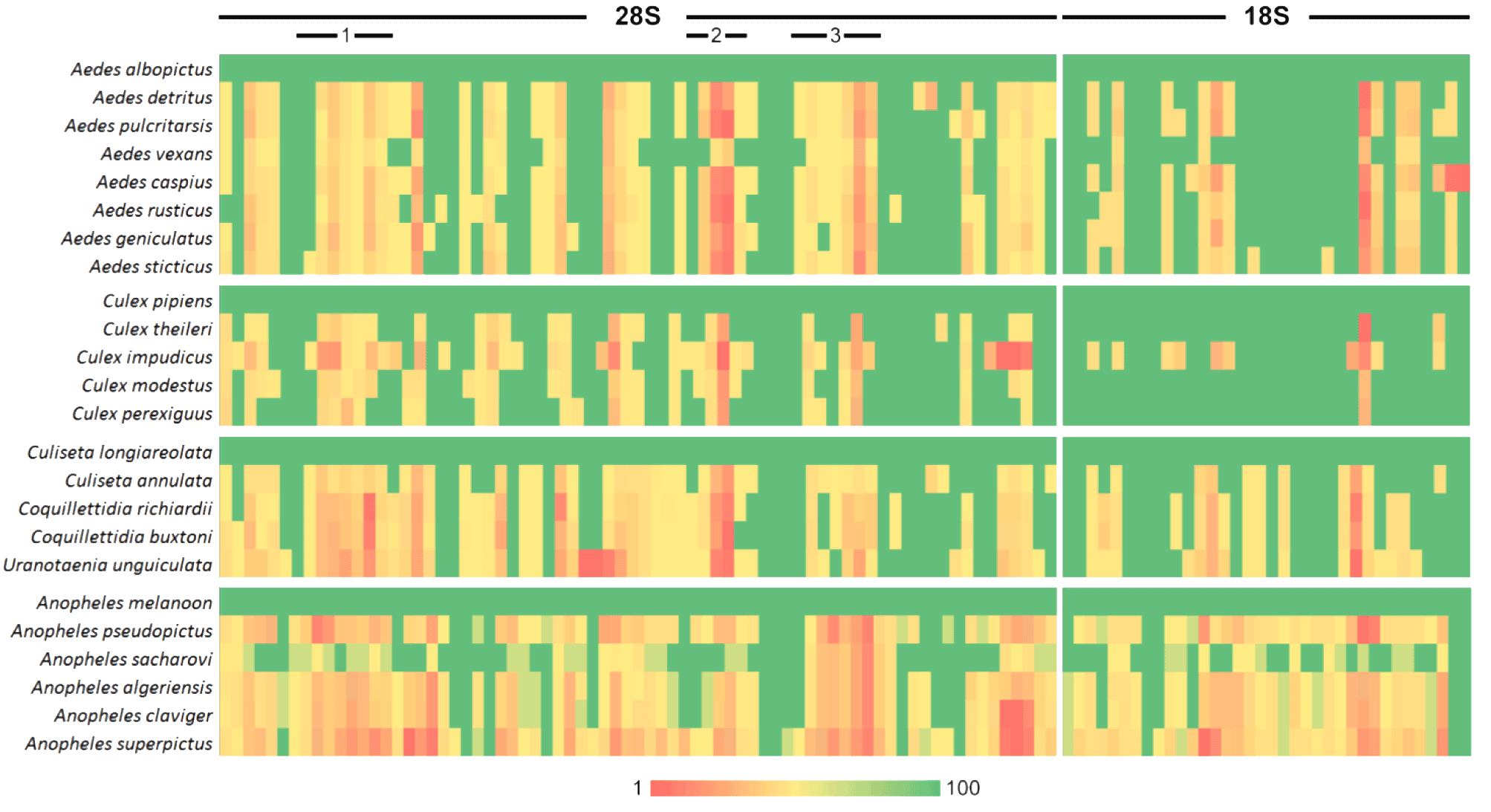
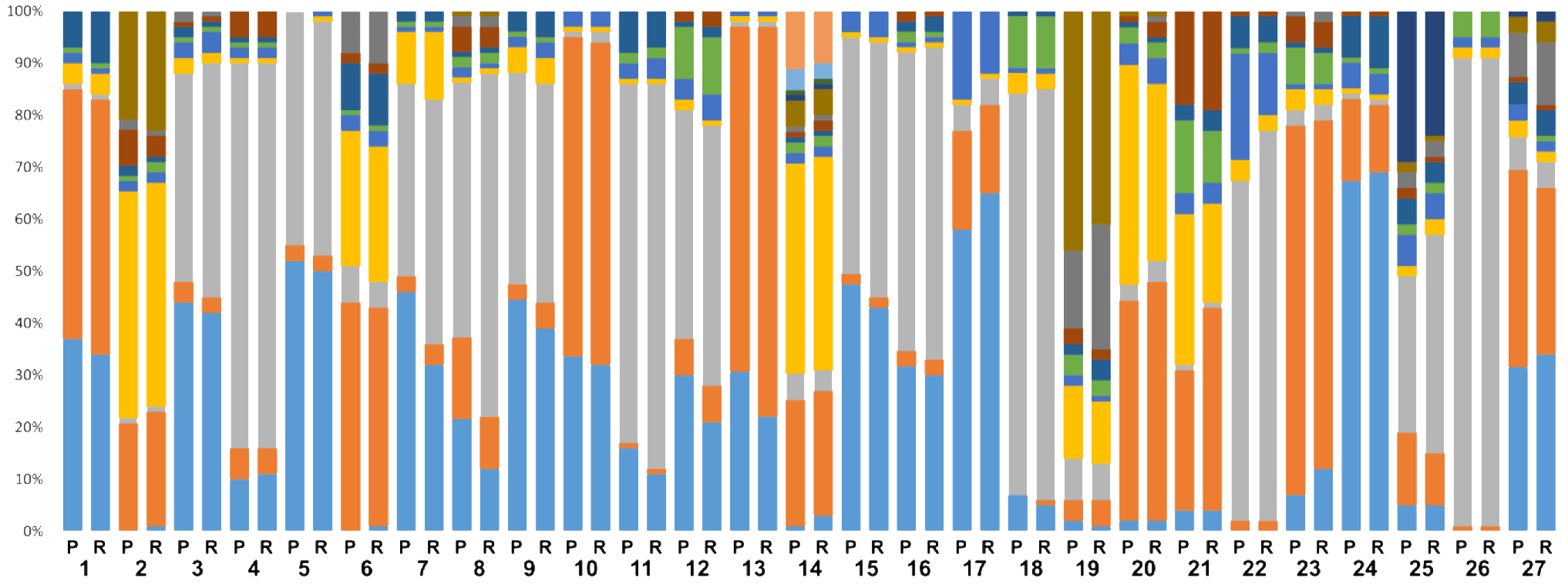
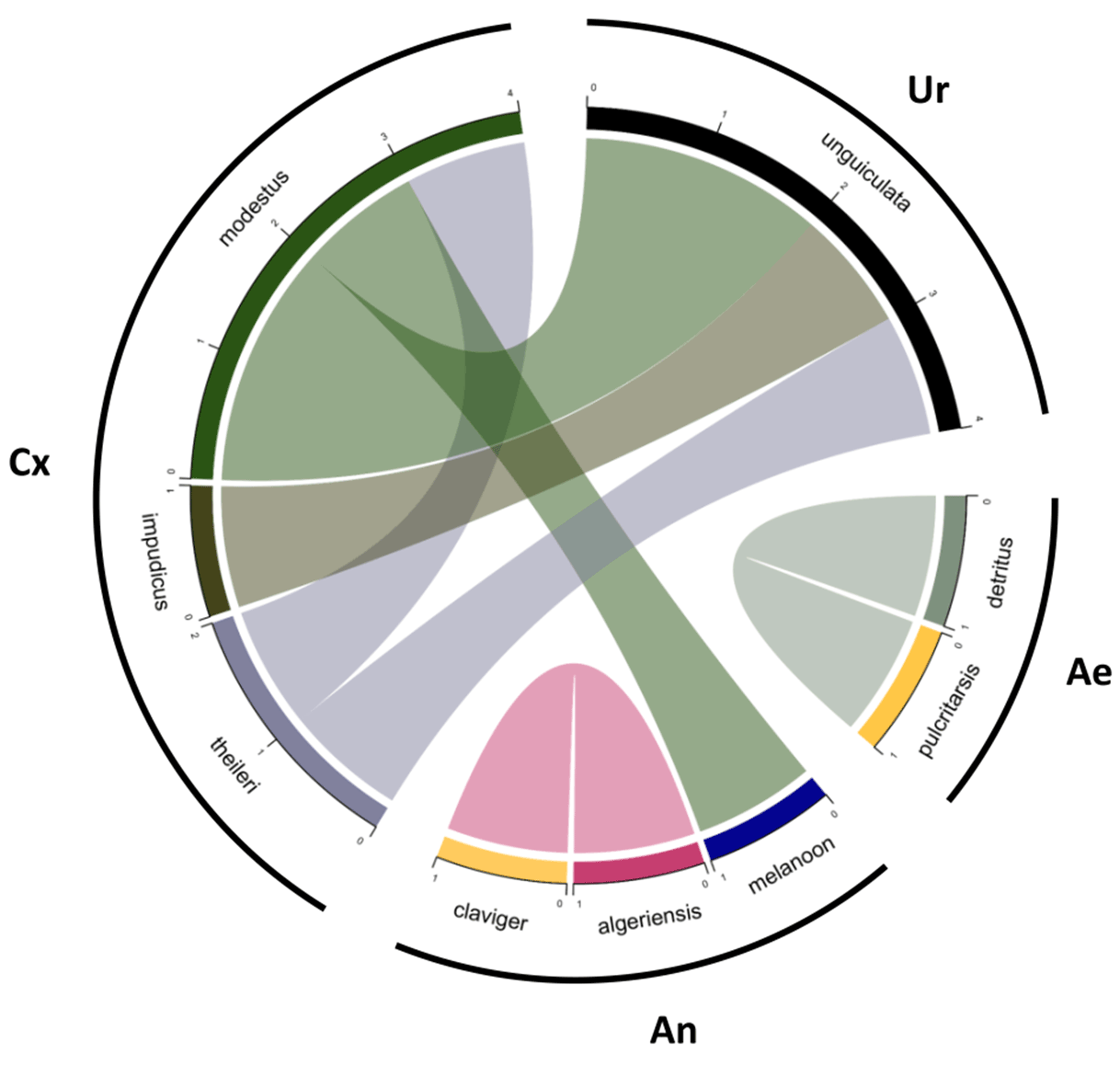
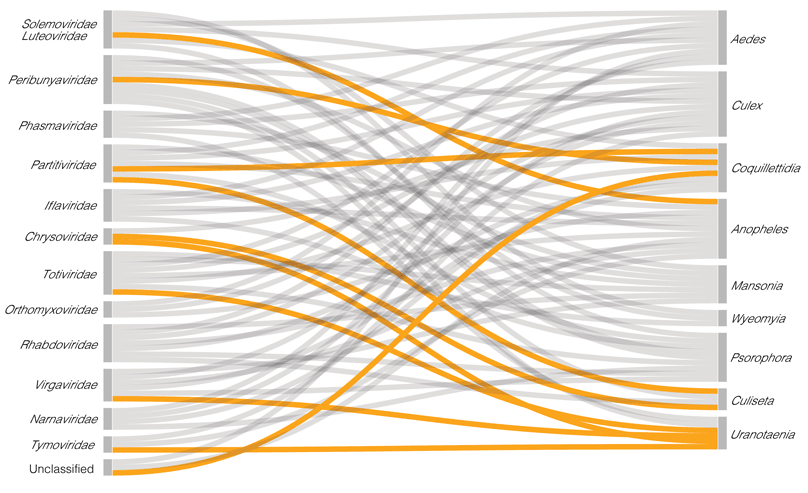
- Konstantinidis, K., Dovrolis, N., Kouvela, A., Kassela, K., Rosa Freitas, M. G., Nearchou, A., De Courcy Williams, M., et al. (2022). ‘Defining virus-carrier networks that shape the composition of the mosquito core virome of a local ecosystem’, Virus Evolution, 8/1. Oxford University Press. DOI: 10.1093/ve/veac036
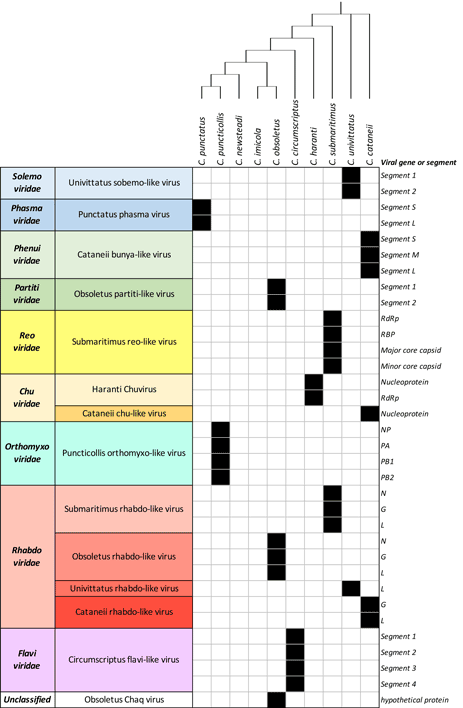
- Konstantinidis, K., Bampali, M., de Courcy Williams, M., Dovrolis, N., Gatzidou, E., Papazilakis, P., Nearchou, A., et al. (2022). ‘Dissecting the Species-Specific Virome in Culicoides of Thrace’, Frontiers in Microbiology, 13. Frontiers Media S.A. DOI: 10.3389/fmicb.2022.802577
- Ridgway, H., Moore, G. J., Mavromoustakos, T., Tsiodras, S., Ligielli, I., Kelaidonis, K., Chasapis, C. T., et al. (2022). ‘Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2’, Computational and Structural Biotechnology Journal, 20: 2091–111. Elsevier B.V. DOI: 10.1016/j.csbj.2022.04.010
- Stefanou, I. K., Dovrolis, N., Gazouli, M., Theodorou, D., Zografos, G. K., & Toutouzas, K. G. (2022). ‘MiRNAs expression pattern and machine learning models elucidate risk for gastric GIST’, Cancer Biomarkers, 33/2: 237–47. IOS Press BV. DOI: 10.3233/CBM-210173
- Talli, I., Dovrolis, N., Oulas, A., Stavrakaki, S., Makedou, K., Spyrou, G. M., & Maroulakou, I. (2022). ‘Novel clinical, molecular and bioinformatics insights into the genetic background of autism’, Human Genomics, 16/1. BioMed Central Ltd. DOI: 10.1186/s40246-022-00415-x
- Vrazas, V., Moustafa, S., Makridakis, M., Karakasiliotis, I., Vlahou, A., Mavromara, P., & Katsani, K. R. (2022). ‘A Proteomic Approach to Study the Biological Role of Hepatitis C Virus Protein Core+1/ARFP’, Viruses, 14/8. MDPI. DOI: 10.3390/v14081694
2021
- Mpekoulis, G., Frakolaki, E., Taka, S., Ioannidis, A., Vassiliou, A. G., Kalliampakou, K. I., Patas, K., et al. (2021). ‘Alteration of L-Dopa decarboxylase expression in SARS-CoV-2 infection and its association with the interferon-inducible ACE2 isoform’, PLoS ONE, 16/6 June. Public Library of Science. DOI: 10.1371/journal.pone.0253458
- Ntolios, P., Tzilas, V., Bouros, E., Avdoula, E., Karakasiliotis, I., Bouros, D., & Steiropoulos, P. (2021). ‘The role of microbiome and virome in idiopathic pulmonary fibrosis’, Biomedicines, 9/4. MDPI AG. DOI: 10.3390/biomedicines9040442
- Andreou, N.-P., Legaki, E., Dovrolis, N., Boyanov, N., Georgiou, K., Gkouskou, K., & Gazouli, M. (2021). ‘B-cell activating factor (BAFF) expression is associated with Crohn’s disease and can serve as a potential prognostic indicator of disease response to Infliximab treatment’, Digestive and Liver Disease, 53/5: 574–80. Elsevier B.V. DOI: 10.1016/j.dld.2020.11.030
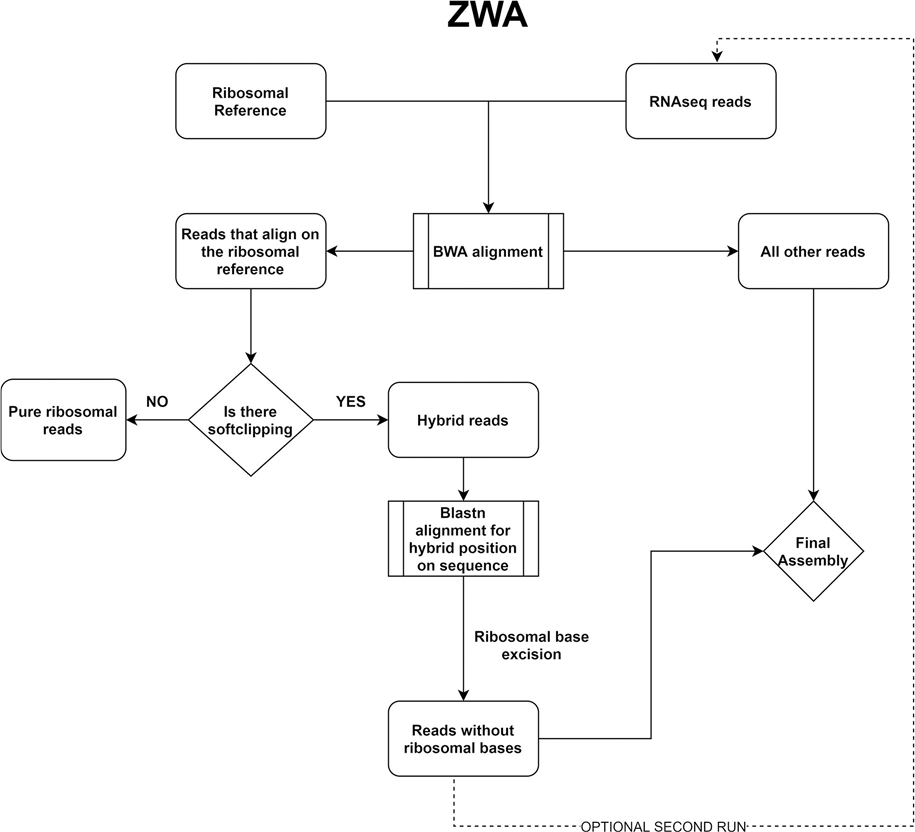
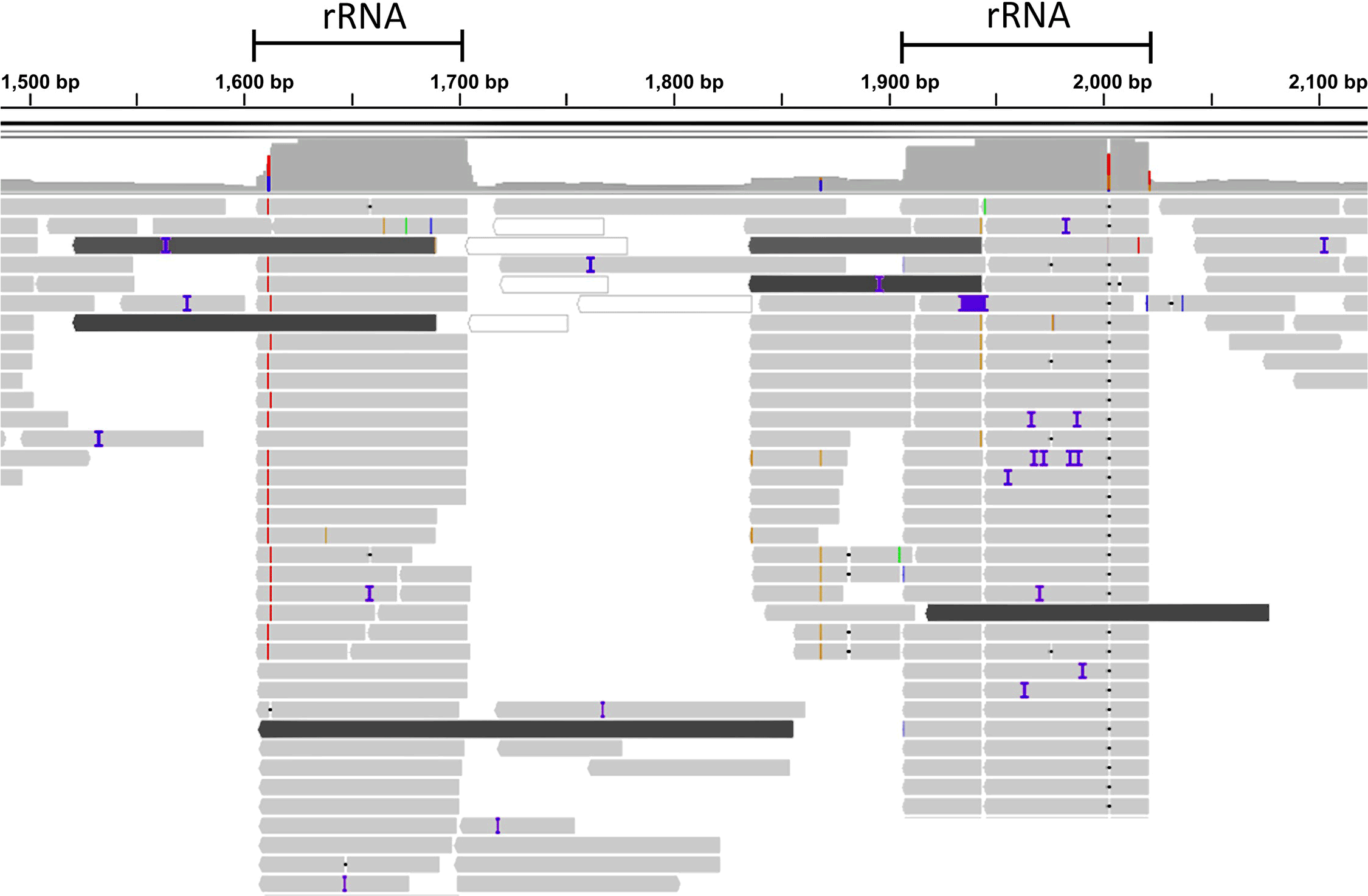
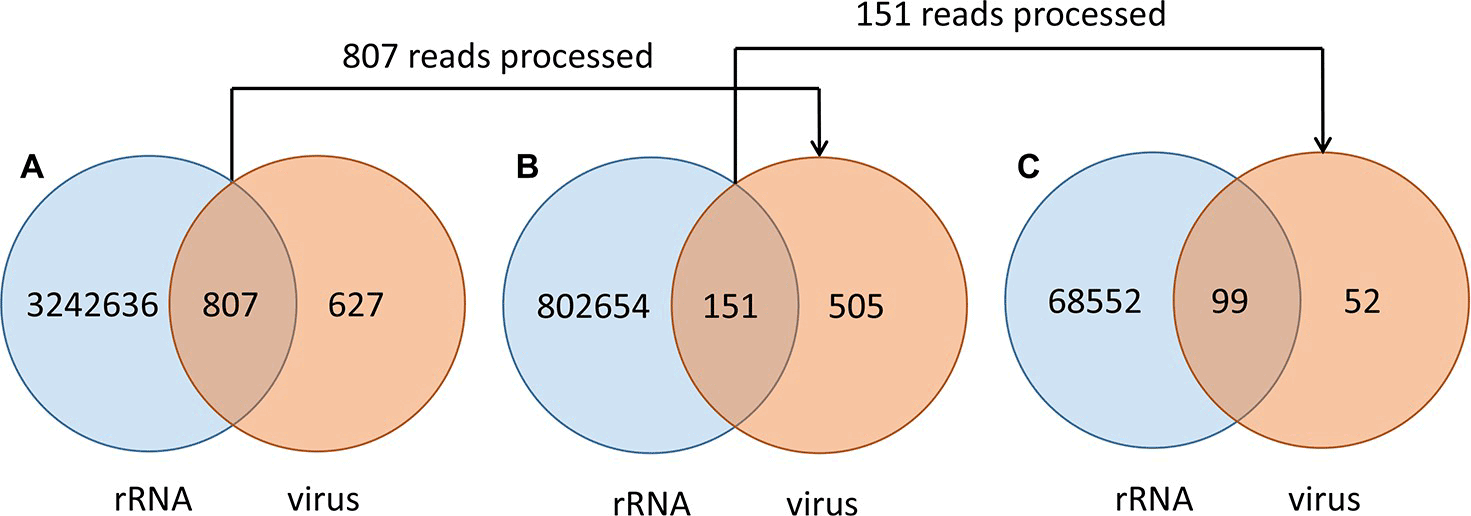
- Dovrolis, N., Kassela, K., Konstantinidis, K., Kouvela, A., Veletza, S., & Karakasiliotis, I. (2021). ‘ZWA: Viral genome assembly and characterization hindrances from virus-host chimeric reads; a refining approach’, PLoS Computational Biology, 17/8. Public Library of Science. DOI: 10.1371/journal.pcbi.1009304
- Spanakis, N., Kassela, K., Dovrolis, N., Bampali, M., Gatzidou, E., Kafasi, A., Froukala, E., et al. (2021). ‘A main event and multiple introductions of SARS-CoV-2 initiated the COVID-19 epidemic in Greece’, Journal of Medical Virology, 93/5: 2899–907. John Wiley and Sons Inc. DOI: 10.1002/jmv.26778
- Konstantinidis, K., Karakasiliotis, I., Anagnostopoulos, K., & Boulougouris, G. C. (2021). ‘On the estimation of the molecular inaccessible volume and the molecular accessible surface of a ligand in protein-ligand systems’, Molecular Systems Design and Engineering, 6/11: 946–63. Royal Society of Chemistry. DOI: 10.1039/d1me00053e
- Karakasiliotis, I., Lagopati, N., Evangelou, K., & Gorgoulis, V. G. (2021). ‘Cellular senescence as a source of SARS-CoV-2 quasispecies’, FEBS Journal. John Wiley and Sons Inc. DOI: 10.1111/febs.16230
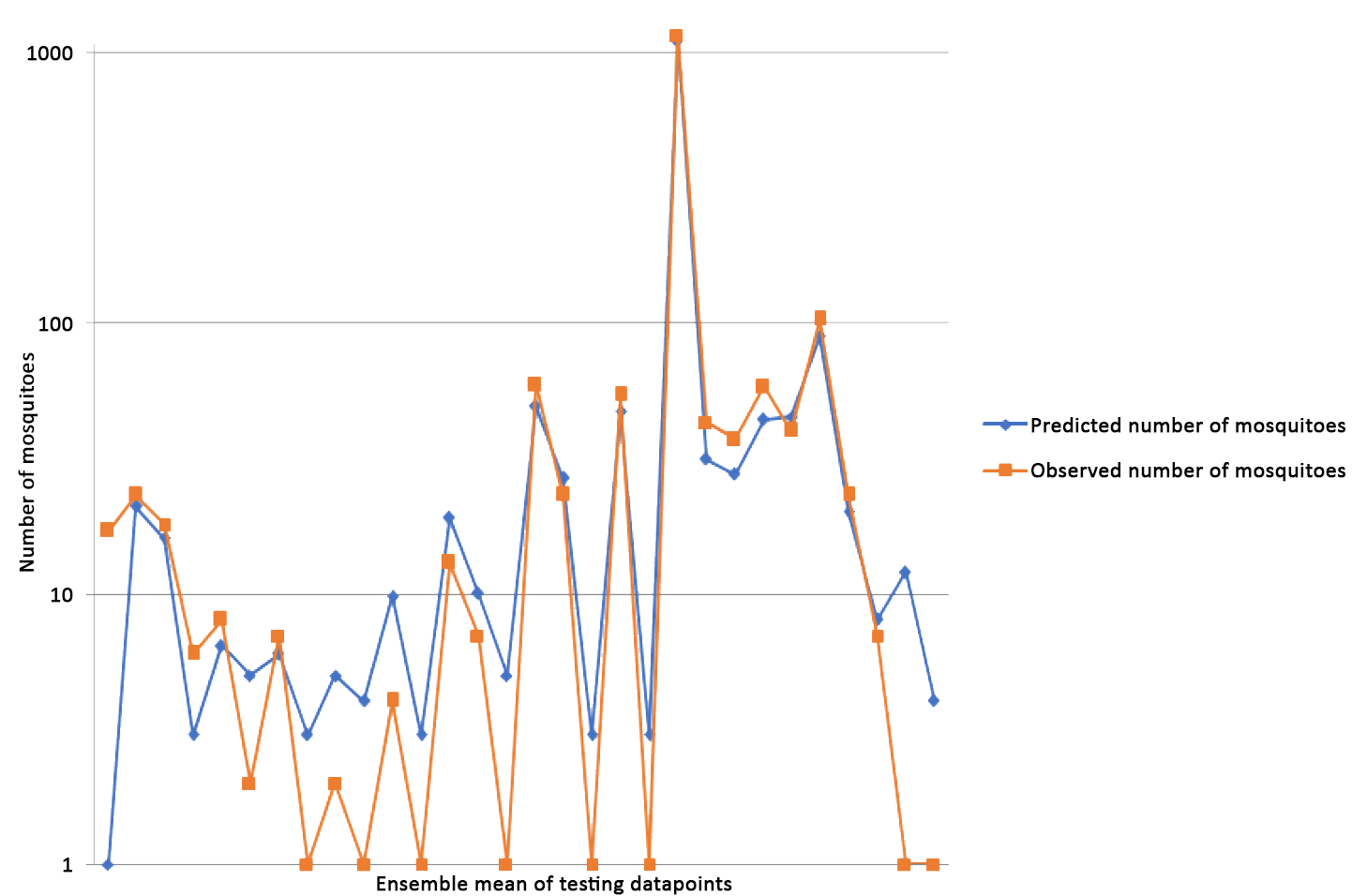
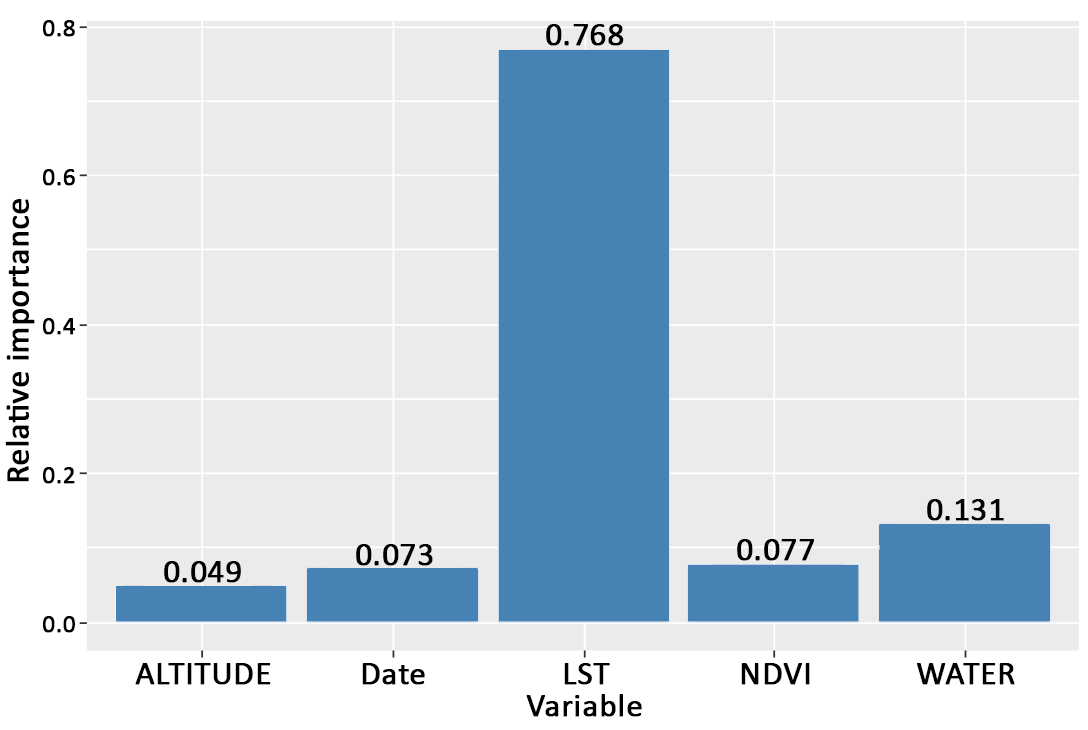
- Kofidou, M., Williams, M. C., Nearchou, A., Veletza, S., Gemitzi, A., & Karakasiliotis, I. (2021). ‘Applying remotely sensed environmental information to model mosquito populations’, Sustainability (Switzerland), 13/14. MDPI AG. DOI: 10.3390/su13147655
2019
- Kassela, K., Karakasiliotis, I., Kokkiou, E., Souvalidou, F., Mimidis, P., Veletza, S., Panopoulou, M., et al. (2019). ‘Intergenotypic 2k/1b hepatitis C virus recombinants in the east macedonia and thrace region of Greece’, Annals of Gastroenterology, 32/1: 88–92. Hellenic Society of Gastroenterology. DOI: 10.20524/aog.2018.0322
2018
- Christodoulou-Vafeiadou, E., Ioakeimidis, F., Andreadou, M., Giagkas, G., Stamatakis, G., Reczko, M., Samiotaki, M., et al. (2018). ‘Divergent innate and epithelial functions of the RNA-binding protein HuR in intestinal inflammation’, Frontiers in Immunology, 9/NOV. Frontiers Media S.A. DOI: 10.3389/fimmu.2018.02732
- Moustafa, S., Karakasiliotis, I., & Mavromara, P. (2018). ‘Hepatitis C virus core+1/ARF protein modulates the cyclin D1/pRb pathway and promotes carcinogenesis’, Journal of Virology, 92/9. American Society for Microbiology. DOI: 10.1128/JVI.02036-17
- Mylopoulou, T., Papadopoulos, V., Kassela, K., Karakasiliotis, I., Souvalidou, F., Mimidis, P., Veletza, S., et al. (2018). ‘Relationship between antibodies to hepatitis C virus core+1 protein and treatment outcome’, Annals of Gastroenterology, 31/5: 593–7. Hellenic Society of Gastroenterology. DOI: 10.20524/aog.2018.0290
2017
- Kassela, K., Karakasiliotis, I., Charpantidis, S., Koskinas, J., Mylopoulou, T., Mimidis, K., Sarrazin, C., et al. (2017). ‘High prevalence of antibodies to core+1/ARF protein in HCV-infected patients with advanced cirrhosis’, Journal of General Virology, 98/7: 1713–9. Microbiology Society. DOI: 10.1099/jgv.0.000851